
MDR Certification: EU market access for medical devices
Secure EU market access and navigate Medical Device Regulation (MDR) compliance with confidence through DNV’s expert certification services and global regulatory support.
Journey continues on Veracity, DNV's trusted digital platform.
MDR Certification: EU market access for medical devices
Navigate EU MDR compliance with confidence
The Medical Devices Regulation (EU) 2017/745 (MDR) establishes a robust regulatory framework for medical devices within the European Union. As a designated Notified Body (NB 2460), DNV offers comprehensive support to manufacturers seeking MDR certification, ensuring streamlined access to the EU market. Our comprehensive services are grounded in global experience and strong technical expertise.
We offer global reach with local support, working closely with recognized regulatory bodies. To streamline the process, we provide combined audits for MDSAP, ISO 13485, and EU regulations such as MDR and IVDR, helping minimize audit duration, reduce impact on operations by audit conduction and reduce costs.
What is MDR certification?
The MDR, effective from 26 May 2021, replaces the Medical Device Directive (MDD) and the Active Implantable Medical Device Directive (AIMDD). It introduces enhanced requirements for medical device safety, performance, and post-market surveillance.
Manufacturers must demonstrate compliance through rigorous conformity assessments conducted by a Notified Body like DNV.

Who needs MDR certification?
MDR certification is mandatory for:
- Manufacturers of medical devices intended for the EU market.
- Manufacturers of devices without a medical purpose as identified in annex XVI.
- EU importers and distributors who translate or repackage medical devices after the manufacturer has sold them, and who are not doing this work for the manufacturer.
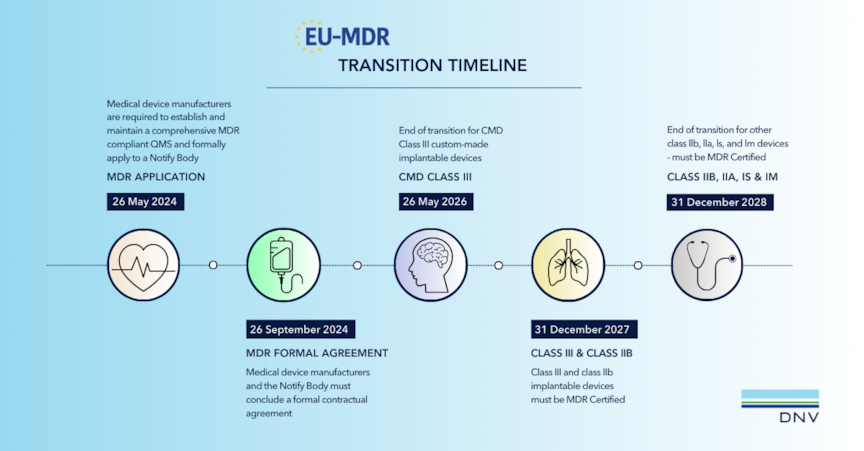
Devices previously certified under MDD or AIMDD must transition to MDR compliance within specified timelines. MDD manufacturers with a valid letter of conformity from a Notified Body may continue transitioning their devices; all others must apply for new MDR certification.
DNV’s MDR certification services
DNV provides a comprehensive suite of services to support your MDR compliance journey:
- Conformity assessment: Evaluation of technical documentation and quality management systems.
- Technical file reviews: Detailed examination of device documentation, including clinical evaluation and risk management.
- Quality systems audits: Initially and throughout the certification cycle for continuous verification of compliance with the requirements.
- Post-market surveillance review: Evaluation of procedures and reports for ongoing device monitoring and reporting.
- Training: Educational resources to help understand MDR requirements.
Steps to achieve MDR certification with DNV
- Application submission: Provide necessary documentation and complete the application process.
- Technical documentation review: DNV assesses your technical files for compliance.
- Quality management system audit: On-site evaluation of your QMS implementation.
- Certification decision: Upon successful assessment, DNV issues the MDR certificate.
- Surveillance activities: Ongoing monitoring to ensure continued compliance
Resources to support your MDR certification journey
Access expert insights, practical guides, and key regulatory references to help you navigate MDR (EU 2017/745) requirements and maintain compliance with confidence.

Medical Device Certification
Step-by-step overview of the MDR certification process and key regulatory obligations for manufacturers.

Application and conformity assessment for medical devices
Guidance on preparing your technical documentation and completing conformity assessments in line with MDR requirements.

List of Standard Fees for MDR Conformity Assessment
Transparent overview of DNV’s fee structure for MDR conformity assessments to help manufacturers plan certification budgets.

Declaration of Interest
Ensure transparency and impartiality throughout the MDR certification process with DNV’s declaration of interest procedure.



